A síndrome de Marfan, também conhecida como Aracnodactilia, é uma desordem do tecido conjuntivo caracterizada por membros anormalmente longos. A doença também afeta outras estruturas do corpo, incluindo o esqueleto, os pulmões, os olhos, o coração e os vasos sangüíneos, mas de maneira menos óbvia. Seu nome vem de Antoine Marfan, o pediatra francês que primeiro a descreveu, em 1896.
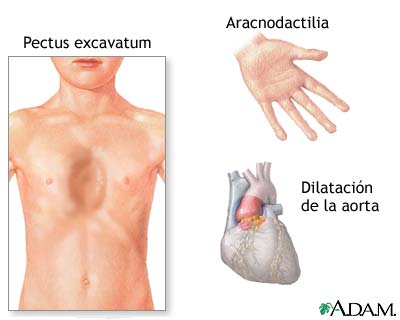
A síndrome de Marfan é uma doença genética associada a deficiências do tecido conjuntivo (desempenha uma função de suporte nos diversos órgãos do corpo). Como resultado, os indivíduos com esta doença apresentam frequentemente anomalias a nível esquelético, ocular e cardiovascular, entre outras. Muitos dos indivíduos afectados têm alterações das válvulas cardíacas e dilatação da aorta. As complicações cardiovasculares mais importantes em termos de risco de vida são os aneurismas da aorta e as dissecções da aorta. A prevealência é de aproximadamente 1 em 5000 indivíduos.
Diagnóstico
As principais manifestações clínicas da Síndrome de Marfan concentram-se em três sistemas principais: o esquelético, caracterizado por estatura elevada, escoliose, braços e mãos alongadas e deformidade torácica; o cardiovascular, caracterizado por alterações da válvula mitral e dilatação da aorta; e o ocular, caracterizado por miopia e luxação do cristalino. Esta capacidade de atingir órgãos tão diferentes denomina-se pleiotropia. Entre as manifestações clínicas, destacam-se as mais potencialmente fatais: os aneurismas da aorta e dissecções da mesma.
A válvula mitral encontra-se localizada entre o ventrículo e o átrio esquerdos e sua função é deixar passar adiante o sangue na direção do ventrículo esquerdo e não permitir seu retorno ou refluxo ao átrio.
Na Síndrome de Marfan, a válvula mitral, freqüentemente, se apresenta alterada, espessada, redundante e com prolapso de parte dela para o interior do átrio esquerdo. Este prolapso pode levar a disfunção importante da válvula com vazamento ou refluxo significativos. Estas alterações são avaliadas através do exame clinico e do ecocardiograma, e também são passiveis de correção cirúrgica. Uma possível complicação do prolapso valvar mitral é a endocardite, ou seja, a infecção bacteriana da própria válvula mitral, motivo pelo que se recomenda a profilaxia para endocardite através da administração de antibióticos antes de qualquer procedimento médico como uma extração dentária ou uma cirurgia.
veja os cuidados com os dentes que você deve ter. são mais suscetíveis a cáries e doença periodontal. O tecido conjuntivo é afetado, provocando alterações na arcada e no palato. Isso afeta diretamente os dentes e suas funções.
O palato (região do céu da boca) em forma de V, provoca apinhamento dental, mordida cruzada e, comumente, desvio do alinhamento normal. Há maior ocorrência de maloclusão classe II.
Os portadores da síndrome de Marfan têm as maxilas estreitas e palato em formato de ogiva. Estes formatos podem criar problemas dentais e ortodônticos. Para uma eficaz correção dos dentes, você deve informar ao dentista que seu filho é portador da síndrome. Os pacientes com prolapso da válvula mitral e/ou válvulas artificiais de coração estão em risco de contrair endocardite(infecção do coração e das válvulas do coração) ao fazer cirurgias dentais. Antes de qualquer procedimento cirúrgico, o médico ou dentista devem ser informados, caso você seja portador da síndrome de Marfan. Assim, você será medicado corretamente. Existem tratamentos profiláticos para endocardite.
Todos os portadores da síndrome devem ter uma correta higiene bucal diária para evitar complicações quanto a endocardite. Os cuidados na prevenção são essenciais no tratamento odontológico.
Um desses cuidados é a antibioticoterapia profilática (tomar antibiótico antes do tratamento odontológico). Cirurgião-Dentista e Cardiologista devem trabalhar em conjunto. É muito importante realizar o maior número possível de intervenções por visita ao consultório a fim de diminuir a ingestão de antibióticos.
A prevenção de cárie e doença periodontal torna-se primordial, desde a boa escovação, uso de fio dental, bochechos com flúor e a troca freqüente de escovas. Isso porque as bactérias se alojam na escova, tornando risco de infecção gengival e até mesmo endocardite.
Tenha em mente que, embora o portador da Síndrome de Marfan necessite de cuidados especiais, nada impossibilita o tratamento odontológico.
A artéria aorta é a maior e principal artéria que sai do coração. Ela conduz o sangue oxigenado para o resto do organismo e com este fim a aorta recebe com cada batimento cardíaco o fluxo sanguíneo proveniente do coração.
No curso da vida a aorta deve acompanhar o crescimento do individuo. Acontece que, na Síndrome de Marfan, devido ao esforço constante a que ela e submetida e a debilidade da parede aórtica decorrente do defeito na fibrilina, a largura da aorta, especialmente na porção inicial pode apresentar um crescimento excessivo, predispondo a várias complicações potencialmente graves, como a dissecção aórtica ou a insuficiência da válvula aórtica (a válvula aórtica forma parte da porção inicial da artéria aorta). Estas alterações aparecem mais freqüentemente a partir da terceira década de vida, porém, podem ocorrer mais precocemente. Eventualmente os pacientes portadores da Síndrome de Marfan necessitam da correção cirúrgica destas complicações. Por este motivo, se não existe contra-indicação, todos os pacientes portadores da Síndrome de Marfan devem receber medicação para diminuir a freqüência e a força dos batimentos cardíacos, no caso, são utilizados os betabloqueadores tais como o atenolol ou propranolol.
Valvula Mitral Marfan
Embora raros, nos casos pode haver também problemas de mal funcionamento na vávula tricúspide e pulmonar
Junto com as limitações de ordem ortopédica, as alterações na aorta constituem-se motivos pelos que a prática de exercícios físicos competitivos é desaconselhada para todos pacientes portadores da Síndrome de Marfan. As principais informações médicas a respeito dão conta que a base genética na maioria dos casos consiste na mutação de um gene situado no cromossomo 15, a fibrilina, importante componente na formação das fibras elásticas. Sua produção anormal resulta em fibras elásticas anormais produzindo as alterações que caracterizam a síndrome. Problemas cárdio-vasculares estão presentes na maioria dos pacientes, sendo a lesão da aorta a principal manifestação. Esta pode apresentar-se em seus primeiros sinais por dilatação da raiz da aorta e do seu anel, no local de origem da artéria no coração, levando à insuficiência aórtica progressiva. Estas alterações favorecem a dilatação das cavidades cardíacas esquerdas, cuja estrutura interna que dá sustentação ao músculo cardíaco é também afetada. A aorta é rica em fibras elásticas. Estas fibras têm uma distribuição uniforme por toda a circunferência da aorta e está entremeada por grande número de fibras musculares e discreta matriz de sustentação. Uma das complicações da síndrome de Marfan é a dissociação de suas estruturas com separação da sua parede em duas paredes finas, formando um lúmen paralelo com desvio e grande prejuízo da circulação. Isto ocorre em 36% dos casos, independente da dilatação aórtica, pondendo apresentar-se como primeira manifestação da doença, sendo mais frequente na porção inicial da aorta (porção ascendente). A valva mitral (estrutura que separa as duas câmaras cardíacas esquerdas) apresenta seus componentes de aspecto alongado e redundante, podendo haver calcificação com endurecimento do anel valvar em 10% dos pacientes. No comprometimento mitral as mulheres são mais freqüentemente afetadas que os homens.
Alterações Cardiovasculares
Coração Marfan A Síndrome de Marfan ocasiona em uma importante parcela dos pacientes problemas na aorta e na válvula mitral. Ambas estruturas possuem uma grande quantidade de fibrilina, proteína defeituosa na síndrome. A fibrilina forma parte das fibras elásticas e contribui na sustentação da parede aórtica e da válvula mitral, conseqüentemente todo paciente portador da Síndrome de Marfan deve ser avaliado periodicamente por um cardiologista, na procura de alterações, em especial na aorta e na válvula mitral.
Genes estudados
FBN1 (fibrillina-1). O síndrome de Marfan clássico é causado por mutações no gene FBN1 (fibrilina-1). Estas mutações podem ser encontradas em 90% dos indivíduos que preenchem os critérios de diagnóstico clínico. Alternativamente, foram descritas mutações nos genes TGFBR2 e TGFBR1 nestes doentes. No caso destes dois genes, o quadro clínico pode ser um pouco diferente e inclui o síndrome de Marfan tipo 2 e o síndrome de Loeys-Dietz (aneurisma aórtico). A proteína em falta (codificada pelo gene FBN1) é a fibrilina que, quando ausente leva a que os ligamentos e as artérias se tornem mais flácidos e frágeis. A flacidez nos ligamentos articulares provoca uma hipermobilidade articular e a uma perda de contenção do crescimento dos ossos. Desta forma, eles crescem demais e deformam-se. Por sua vez, o ligamento de sustentação do cristalino (zónula), que neste síndrome é frágil, leva à mobilidade da lente (subluxação) podendo até romper-se (luxação). No coração, o sangue que é bombeado com muita força a cada batimento cardíaco é ejectado directamente na aorta, cujas paredes estão fragilizadas, pelo que esta se vai dilatando (aneurisma da aorta) e pode até chegar a romper-se.
Quadro genético
Através de técnicas de pesquisa do DNA em famílias chegou-se à conclusão de que o gene que fabricava a fibrilina estava localizado no cromossomo 15 e posteriormente, em 1993, uma pesquisadora brasileira, Dra. Lygia Pereira, conseguiu obter toda a seqüência do gene e dar o nome de fibrilina-1.
Este gene é muito longo, constituído por 65 seqüências codificadoras chamadas éxons. Desde esta data foram identificadas mais de 200 mutações diferentes, entre elas as relatadas por um estudo brasileiro concluído em 1997 pela Dra. Ana Beatriz A . Perez. Apesar dos esforços pouco se descobriu sobre a correlação entre as diferentes mutações e as manifestações clínicas. Até o momento sabe-se que mutações no meio do gene causam uma forma da doença muito grave, chamada até de letal porque a criança já nasce com dilatação da aorta e demais características. Na região final do gene as mutações parecem correlacionar-se com quadros clínicos leves. Além disso, as mutações são quase sempre exclusivas de uma determinada família.
O estudo do gene permitiu também aos pesquisadores a constatação de que quadros parecidos e muitas vezes caracterizados por uma única manifestação clínica apresentavam mutação no gene fibrilina-1. Nesse momento nasceu o conceito das “fibrilinopatias”, ou seja, doenças causadas por mutações no gene fibrilina-1, que podem ter uma combinação diferente dos sintomas da Síndrome de Marfan clássica assim como estar associadas com manifestações menos comuns ou não relacionadas a princípio.
Incidência
A incidência deste síndrome é de 1 em 10000 natos-vivos. Trata-se de uma doença de hereditariedade autossómica dominante, no entanto 15% dos casos são mutações de novo. A causa são mutações do gene responsável pela síntese de fibrilina-1, que se encontra localizado no cromossoma 15. A grande dimensão deste gene e a multiplicidade das mutações possíveis não permitem actualmente o diagnóstico molecular de rotina.
Os critérios de diagnóstico de certeza nos indivíduos com história familiar da doença requerem uma manifestação major para o diagnóstico. Naqueles em que não existem antecedentes familiares, pelos menos duas alterações major e o envolvimento de outro sistema devem estar presentes para um diagnóstico correcto.
Evolução
Quanto mais precoces forem as manifestações clínicas, particularmente as do foro cardíaco (presentes em 83% dos doentes), mais reservado é o prognóstico.
Os casos mais característicos são diagnosticados em crianças mais velhas/adolescentes e as complicações mais frequentes são as oculares (luxação do cristalino, 70%, miopia, 60%), as cardíacas (dilatação do arco aórtico, 84%, prolapso da válvula mitral, 58%) e as do foro ósseo (escoliose, 44%, tórax côncavo, 68%, pé plano, 44%). A maioria destes indivíduos são altos (56% têm estatura> P95 para a idade) e têm aracnodactilia (88%).
A sintomatologia cardíaca relacionada com o prolapso da válvula mitral inclui palpitações e dispneia. A dor torácica pode ter implicações mais graves, nomeadamente quando associada ao pneumotórax ou a dissecção da aorta. Ambas as situações carecem de intervenção urgente. A terapêutica médica com beta-bloqueantes é eficaz na prevenção da patologia aórtica.
Outros problemas associados são:
- sobreposição dentária e palato alto;
- doença pulmonar restritiva por escoliose severa;
- predisposição para cataratas;
- problemas da retina, incluindo descolamento;
- predisposição para equimoses fáceis, com cicatrização normal;
- dor na nuca e abdómen por alongamento da medula espinhal;
- lesões dos ligamentos, artralgias e fracturas em 96% dos adultos (raro antes dos 5 anos de idade);
- gravidez complicada por osteoporose e pelo risco aumentado de dissecção da aorta.
Tratamento e alertas
Embora não haja nenhuma cura para a síndrome de Marfan, há muitas opções para controlar os sintomas a circunstância. Porque a síndrome de Marfan é raro, é importante encontrar um médico que seja conhecedor sobre a circunstância. Durante a examinação física inicial, um historiam médico detalhado e de família será feito exame junto com a medida da altura, o exame do olho e um electrocardiograma. Uma avaliação esquelética anual para detectar todas as mudanças na coluna ou no esterno é conduzida tipicamente. Esta avaliação é particularmente importante durante o período de elevado crescimento do adolescente. Uma anomalia grave não só desfigura mas pode também impedir que o coração e os pulmões funcionem correctamente. Em alguns casos, uma cinta ou uma cirurgia ortopédica podem ser recomendadas limitar os danos. As examinações regulares do olho são vitais para descobrir e corrigir todos os problemas da visão. Em a maioria de casos os vidros ou as lentes de contacto podem corrigir o problema. Entretanto, a cirurgia pode ser necessária em alguns casos. Análises regulares e os ecocardiogramas ajudam a avaliar o tamanho da aorta e a maneira que o coração está trabalhando. Um problema potencial é identificado mais cedo e tratado, mais baixo o risco de complicações que ameacem a vida. Aqueles com problemas do coração devem usar uma pulseira de aviso médico e ir às emergências se sentirem dores no peito, costas, ou abdominal. Alguns problemas da válvula do coração podem ser controlados com medicamentos, tais como o betablocante, que pode ajudar diminuir o stress na aorta. Entretanto, a cirurgia para substituir uma válvula ou para reparar a aorta pode ser necessária.
Os pacientes de Marfan geralmente são medicados com beta-bloqueantes de forma a diminuir a frequência cardíaca e o inotropismo do coração (força de contracção). Em caso de aneurisma da aorta, recorre-se a intervenção cirúrgica. É ainda importante que o doente seja alertado para a importância de uma correcta higiene oral, de forma a prevenir a entrada de bactérias por essa via e evitar assim complicações como endocardites.
Um medicamento frequentemente prescrito para tratar a Hipertensão Arterial evitaria as graves complicações - às vezes mortais - da Síndrome de Marfan, uma doença genética que debilita a estrutura dos vasos sanguíneos, segundo um estudo norte-americano publicado na Science. As pessoas que sofrem desta anomalia correm um risco elevado de desenvolver um Aneurisma da Aorta que pode levar a mortes súbitas. O estudo, realizado em cobaias pelo Howard Hughes Medical Institute da Johns Hopkins University, EUA, mostrou que o fármaco, losartan atenua a progressão da Síndrome de Marfan. O tratamento também permitiu reparar a estrutura da parede da aorta. "Depois do tratamento com losartan, as cobaias apresentavam um crescimento normal do canal aórtico, (...) bem como uma espessura normal da parede da aorta", destacou Harry Dietz, investigador que liderou o estudo. Em resumo, depois da toma deste medicamento não houve diferenças entre os animais estudados e os considerados normais, segundo os resultados publicados na revista Science. O losartan dificulta o desenvolvimento do Aneurisma ao reduzir a actividade de uma molécula denominada factor de transformação celular beta. Os cientistas descobriram recentemente que esta molécula é, muito provavelmente, a responsável pelas graves carências geradas pela Síndrome de Marfan.
Aspectos psicológicos
Deformidades anatômicas e fisiológicas, características em portadores da síndrome de Marfan, são fatores que provocam distúrbio de ajustamento psicológico com limitações no desenvolvimento emocional e social. A aparência física pouco comum nesses indivíduos freqüentemente leva a alterações no esquema corporal, na auto-estima e, conseqüentemente, no desenvolvimento global da personalidade. As deformidades do corpo são responsáveis por colocar o indivíduo em uma posição de destaque negativo junto ao grupo, causando prejuízos muitas vezes determinantes na vida da pessoa tais como, inibição, sentimentos de menosvalia e tendência ao isolamento. Por ser uma doença crônica e peculiar, atingindo precocemente o indivíduo, a síndrome pode provocar danos na área afetivo-relacional em cada etapa do desenvolvimento.
Na infância, como na maioria das cardiopatias congênitas, a síndrome de Marfan pode estar freqüentemente associada a distúrbios de aprendizagem decorrentes de deficit cognitivos provocados pela doença ou ainda problemas emocionais diversos. As limitações físicas podem impedir a criança de ter acesso a muitas formas de brincadeiras e exercícios, reduzindo assim sua capacidade de expressão e estruturação da personalidade adquiridas com estas atividades.
Na puberdade e adolescência, período crítico do desenvolvimento, o portador da síndrome defronta-se com a superposição de conflitos relacionados às transformações corporais e psíquicas.
No adulto as dificuldades não são menos traumáticas devido a muitos fatores ansiogênicos que acompanham o quadro, tais como sentimento de insegurança quanto a ser aceito pelo parceiro do sexo oposto, necessidade de planejamento familiar. No caso da mulher, pode haver interdição relativa para a gestação, o que implicaria a perda da integridade e o luto pela renúncia à maternidade, ou então a experiência da gravidez de alto risco, também ansiogênica. A reduzida expectativa de vida em ambos os sexos é outro fator causador de danos emocionais como os variados graus de estados depressivos, limitando a realização de projetos pessoais e profissionais.
Estudos apontaram significativo ônus nas atividades físicas diárias, na educação formal e na oportunidade de emprego. A auto-imagem, afetada pela aparência, tem repercussões negativas no comportamento social, caracterizado por introversão.
As principais manifestações oculares em portadores da síndrome de Marfan são:subluxação do cristalino, miopia e descolamento de retina.
Subluxação do cristalino é o deslocamento do cristalino da sua posição normal no eixo visual. O descolamento de retina representa a complicação ocular mais grave desta síndrome.
Algumas manifestações oculares são mais frequentes em pacientes com síndrome de Marfan quando comparados com a população geral, dentre elas: estrabismo, catarata e glaucoma.
Fisioterapia
O tratamento fisioterapêutico é parte integrante da equipe interdisciplinar que visa o auxílio terapêutico nos pacientes portadores da Síndrome de Marfan. A atuação deve ser considerada de fundamental importância uma vez que possui atuação em diversos sistemas tais como locomotor, cardiovascular e respiratório.
A atuação quanto ao aparelho locomotor baseia-se em uma terapêutica que tem como objetivo minimizar as deformidades esperadas nestes pacientes tais como cifose, escoliose, alteração da caixa torácica e aracnodactilia. A hipermobilidade das articulações faz com que estes indivíduos sejam mais propensos a desenvolver lesões e luxações, situação esta que também deve ser associada ao processo preventivo e curativo com a terapêutica fisioterápica.
No sistema cardiovascular a fisioterapêutica exerce papel no que diz respeito à melhora do condicionamento cardiorrespiratório e musculoesquelético, através da realização de exercícios planejados e sob monitorização continua, uma vez que estes pacientes podem ser classificados como indivíduos de risco. Normalmente se dá preferência a atividades do tipo aeróbias e de baixa intensidade, impedindo-se a associação com exercícios que ofereçam impacto ou componente isométrico.
Os pacientes com Síndrome de Marfan apresentam deformidades e limitação crônica da caixa torácica, tendo maior probabilidade ao desenvolvimento de doenças respiratórias restritivas. As principais complicações pulmonares são; bronquiectasia, bolhas enfizematosas, pneumotórax espontâneo e fibrose pulmonar. O acompanhamento fisioterapêutico nesses pacientes é de suma importância visando a manutenção dos volumes e capacidades pulmonar.
A fisioterapia atua de forma imprescindível no pré e pós-operatório dos pacientes eletivos de cirurgias ortopédicas e cardiovasculares, diminuíndo o número de complicações no pós–operatório e conseqüentemente o tempo de internação.
Pessoas afetadas
Entre pessoas famosas, acredita-se que tinham a síndrome Júlio César, Charles de Gaulle, Sergei Rachmaninoff, Maria I da Escócia, Abraham Lincoln, o violinista Niccolò Paganini e possivelmente Charles Maurice de Talleyrand. Um livro recente sugere que o faraó egípcio Amen-hotep IV (Akhenaton) pode ter tido esta condição. Alega-se que a colunista política norte-americana Ann Coulter sofre da Síndrome de Marfan. Rumores dizem que Osama bin Laden também tem a Síndrome. [1] [2]
O astro do vôlei Flo Hyman, conhecido por ter Marfan, e o compositor de musicais Jonathan Larson, que acredita-se que também tinha a Síndrome, morreram de dissecção de aorta. O ator Brent Collins, da série de televisão Another World, era um anão com Síndrome de Marfan; aos 46 anos de idade, ele teve um repentino crescimento, o que o levou à morte. O ator Vincent Schiavelli possuía a Síndrome de Marfan, e era um membro honorário da National Marfan Foundation [3]. Também o ateleta Michael Phelps posui este tipo de sindrome[carece de fontes].
No Brasil, já existe a Fundação Marfan Brasil [4] que tem ajudado no avanço das pesquisas sobre a Síndrome de Marfan.
Um comentário:
Casinos Near Me - MapYRO
Search by area and 구미 출장마사지 find the best casinos in Las Vegas, 원주 출장마사지 NV. See what casinos are and search 서울특별 출장안마 for 양산 출장안마 non-stop fun! Find your answer now. 제주 출장안마
Postar um comentário